Yleisyys
Mitokondrio -DNA tai mtDNA on deoksiribonukleiinihappo, joka asuu mitokondrioiden eli eukaryoottisolujen organellien sisällä, jotka ovat vastuussa erittäin tärkeästä hapettavan fosforylaation soluprosessista.

Sillä on kuitenkin myös joitakin rakenteellisia ja toiminnallisia erityispiirteitä, jotka tekevät siitä ainutlaatuisen lajissaan. Näitä erityispiirteitä ovat: nukleotidien kaksoisjuovan pyöreys, geenien sisältö (joka on vain 37 elementtiä) ja ei-koodaavien nukleotidisekvenssien lähes täydellinen puuttuminen.
Mitokondrio -DNA: lla on perustavanlaatuinen tehtävä solujen selviytymisessä: se tuottaa entsyymejä, jotka ovat välttämättömiä hapettavan fosforylaation toteuttamiseksi.
Mikä on mitokondrioiden DNA?
Mitokondrio -DNA tai mtDNA on mitokondrioissa sijaitseva DNA.
Mitokondriot ovat eukaryoottisille organismeille tyypillisiä suuria soluorganelleja, jotka muuttavat elintarvikkeiden sisältämän kemiallisen energian ATP: ksi, joka on solujen hyödyntämä energiamuoto.
MITOCHONDRONSIN RAKENNEEN JA TOIMINNAN TAUSTA
Putkimainen, rihmainen tai rakeinen muoto, mitokondriot sijaitsevat sytoplasmassa, ja ne vievät lähes 25% jälkimmäisen tilavuudesta.
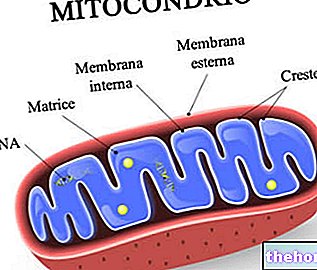
Niissä on kaksi kaksikerroksista fosfolipidikalvoa, toinen ulompi ja toinen sisäinen.
Ulompi kalvo, joka tunnetaan nimellä ulompi mitokondrioiden kalvo, edustaa kunkin mitokondrion kehää ja siinä on kuljetusproteiineja (poriinit ja enemmän), jotka tekevät siitä läpäisevän molekyylejä, joiden koko on enintään 5000 daltonia.
Sisempi kalvo, joka tunnetaan nimellä sisäinen mitokondrioiden kalvo, sisältää kaikki entsymaattiset (tai entsymaattiset) ja koentsyymikomponentit, jotka ovat välttämättömiä ATP: n synteesille, ja se määrittelee keskustilan, jota kutsutaan matriisiksi.
Toisin kuin uloin kalvo, sisäisessä mitokondrio -kalvossa on lukuisia invaginaatioita - niin sanottuja harjanteita - jotka lisäävät sen kokonaispinta -alaa.
Kahden mitokondriaalisen kalvon välissä on lähes 60-80 Angströmia (A). Tätä tilaa kutsutaan kalvojen väliseksi tilaksi. Kalvojen välisen tilan koostumus on hyvin samanlainen kuin sytoplasman.
ATP: n synteesi, jota hoitaa mitokondriot, on hyvin monimutkainen prosessi, jonka biologit tunnistavat termillä oksidatiivinen fosforylaatio.
MITOCHONDRAL DNA: N TARKKA SIJAINTI JA MÄÄRÄ

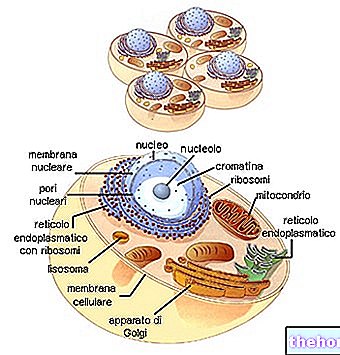
Kuva: ihmisen mitokondrio.
Mitokondrio -DNA sijaitsee mitokondrio -matriisissa, eli sisäisen mitokondriaalisen kalvon rajaamassa tilassa.
Luotettavien tieteellisten tutkimusten mukaan jokainen mitokondrio voi sisältää 2–12 kopiota mitokondrioiden DNA: sta.
Kun otetaan huomioon se tosiasia, että jotkut solut voivat ihmiskehossa sisältää useita tuhansia mitokondrioita, mitokondrioiden DNA: n kopioiden kokonaismäärä yhdessä ihmisen solussa voi nousta jopa 20 000 yksikköön.
Huomaa: mitokondrioiden määrä ihmissoluissa vaihtelee solutyypin mukaan. Esimerkiksi maksasolut (eli maksasolut) voivat sisältää 1 000 - 2 000 mitokondriaa, kun taas punasolut (eli punasolut) ovat täysin vailla niitä.
Rakenne
Mitokondrioiden DNA -molekyylin yleinen rakenne muistuttaa ydin -DNA: n yleistä rakennetta, eli eukaryoottisolujen ytimessä olevaa geneettistä perintöä.
Todellakin, analogisesti ydin -DNA: han:
- Mitokondrio -DNA on biopolymeeri, joka koostuu kahdesta pitkästä nukleotidisäikeestä. Nukleotidit ovat orgaanisia molekyylejä, jotka syntyvät kolmen elementin yhdistämisestä: sokeri, jossa on 5 hiiliatomia (DNA: n tapauksessa, deoksiriboosi), typpipitoinen emäs ja fosfaattiryhmä.
- Kukin mitokondrio -DNA: n nukleotidi sitoutuu saman juosteen seuraavaan nukleotidiin fosfodiesterisidoksen avulla sen deoksiriboosin hiili 3: n ja välittömästi seuraavan nukleotidin fosfaattiryhmän välillä.
- Mitokondrioiden DNA: n kahdella säikeellä on vastakkaiset suuntaukset, ja toisen pää on vuorovaikutuksessa toisen pään kanssa ja päinvastoin.
- Mitokondrio -DNA: n kaksi säiettä ovat vuorovaikutuksessa keskenään typpipitoisten emästen kautta.
Erityisesti kunkin filamentin typpipohjainen emäs muodostaa vetysidoksia yhden ja ainoan typpipitoisen emäksen kanssa, joka on läsnä toisessa filamentissa.
Tämän tyyppistä vuorovaikutusta kutsutaan "pariksi typpeä sisältävien emästen välillä" tai "typpipitoisten emästen pariksi". - Mitokondrioiden DNA: n typpipohjat ovat adeniini, tymiini, sytosiini ja guaniini.
Näiden typpipitoisten emästen muodostama pariliitos ei ole sattumaa, vaan erittäin spesifinen: adeniini on vuorovaikutuksessa vain tymiinin kanssa, kun taas sytosiini on vuorovaikutuksessa vain guaniinin kanssa. - Mitokondrioiden DNA: ssa on geenejä (tai geenisekvenssejä). Geenit ovat enemmän tai vähemmän pitkiä nukleotidisekvenssejä, joilla on hyvin määritelty biologinen merkitys. Useimmissa tapauksissa niistä syntyy proteiineja.

MITOCHONDRAL DNA: n RAKENTEELLISET TIEDOT
Edellä mainittujen analogioiden lisäksi ihmisen mitokondrioiden DNA: lla on joitain rakenteellisia erityispiirteitä, jotka erottavat sen merkittävästi ihmisen ydin -DNA: sta.
Ensinnäkin se on pyöreä molekyyli, kun taas ydin -DNA on lineaarinen molekyyli.
Sillä on siten 16 569 typpipitoista emäsparia, kun taas ydin -DNA: lla on mahtava 3,3 miljardia.
Se sisältää 37 geeniä, kun taas ydin -DNA näyttää sisältävän 20 000 - 25 000 geeniä.
Se ei ole järjestetty kromosomeihin, kun taas ydin -DNA on jaettu 23 kromosomiin ja muodot, joissa on tiettyjä proteiineja, kromatiinia.
Lopuksi se sisältää sarjan nukleotideja, jotka osallistuvat kahteen geeniin samanaikaisesti, kun taas ydin -DNA: ssa on geenejä, joiden nukleotidisekvenssit ovat hyvin määriteltyjä ja erillisiä toisistaan.
Alkuperä
Mitokondrioiden DNA: lla on todennäköisesti "bakteeriperäinen" alkuperä.
Itse asiassa molekyylibiologit uskovat lukuisten riippumattomien tutkimusten perusteella, että mitokondrio -DNA: n solujen läsnäolo on seurausta siitä, että esi -isät eukaryoottisolut ovat sisällyttäneet itsenäisiä bakteeri -organismeja, jotka ovat hyvin samankaltaisia kuin mitokondriot.
Tämä utelias löytö on vain osittain hämmästyttänyt tiedeyhteisöä, koska bakteereissa oleva DNA on yleensä pyöreä nukleotidiketju, kuten mitokondrio -DNA.
Teoria, jonka mukaan mitokondrioilla ja mitokondrioiden DNA: lla on "bakteeriperäisyys", saa nimen "endosymbioottinen teoria" sanasta "endosymbioosi". Lyhyesti sanottuna, biologiassa termi "endosymbioosi" osoittaa kahden organismin yhteistyötä, johon kuuluu mm. "yhdistäminen toiseen, jotta saadaan tietty etu.
Uteliaisuus
Luotettavien tieteellisten tutkimusten mukaan monet tulevassa mitokondrioiden DNA: ssa olevat bakteerigeenit olisivat evoluution aikana muuttaneet sijaintiaan siirtyen ydin -DNA: ksi.
Toisin sanoen endosymbioosin alussa jotkut nyt ydin -DNA: ssa esiintyvät geenit asuivat näiden bakteeri -organismien DNA: ssa, joista myöhemmin tuli mitokondrioita.
Geenien siirtymiseen mitokondrio -DNA: n ja ydin -DNA: n välillä liittyvän teorian tueksi on havainto, että tietyt geenit ovat peräisin mitokondrioiden DNA: sta, joillakin lajeilla, ja ydin -DNA: lla, toisilla.
Toiminto
Mitokondrio -DNA tuottaa entsyymejä (eli proteiineja), joita tarvitaan herkän hapettavan fosforylaatioprosessin oikeaan toteuttamiseen.
Ohjeet näiden entsyymien syntetisoimiseksi ovat 37 geenissä, jotka muodostavat mitokondrioiden DNA -genomin.
MITÄ MITOCHONDRAL DNA GENES KOODI: TIEDOT
Mitokondrioiden DNA: n 37 geeniä koodataan: proteiineille, tRNA: lle ja rRNA: lle.
Erityisesti:
- 13 koodaa 13 proteiinia, jotka ovat vastuussa oksidatiivisen fosforylaation suorittamisesta
- 22 koodi 22 tRNA -molekyylille
- 2 koodaa 2 rRNA -molekyyliä
TRNA- ja rRNA -molekyylit ovat olennaisia edellä mainittujen 13 proteiinin synteesille, koska ne muodostavat koneen, joka säätelee niiden tuotantoa.
Toisin sanoen mitokondrioiden DNA: lla on tietoa tietyn proteiinijoukon tuottamiseksi ja niiden synteesiin tarvittavat työkalut.
Mitä ovat RNA, tRNA ja rRNA?
RNA tai ribonukleiinihappo on nukleiinihappo, jolla on keskeinen rooli proteiinien muodostumisessa DNA: sta alkaen.
Yleensä yksijuosteinen ANN voi olla eri muodoissa (tai tyypeissä) riippuen siitä, mihin toimintoon se on delegoitu.
TRNA ja rRNA ovat kaksi näistä mahdollisista muodoista.
TRNA: ta käytetään aminohappojen lisäämiseen proteiinien valmistusprosessin aikana. Aminohapot ovat molekyyliyksiköitä, jotka muodostavat proteiineja.
RRNA muodostaa ribosomit eli solurakenteet, joissa proteiinien synteesi tapahtuu.
Jos haluat tietää yksityiskohtaisesti ANN: n ja sen toiminnot, lukijat voivat napsauttaa tästä.
TOIMINNALLISET TIEDOT MITOCHONDRAL DNA: sta
Toiminnalliselta kannalta mitokondrio -DNA: lla on joitain erityisiä ominaisuuksia, jotka erottavat sen selvästi ydin -DNA: sta.
Tässä ovat nämä erikoiset ominaisuudet:
- Mitokondrioiden DNA on puoliksi itsenäinen siinä mielessä, että se tarvitsee joidenkin ydin-DNA: sta syntetisoitujen proteiinien väliintuloa.
Toisaalta ydin -DNA on täysin itsenäinen ja tuottaa itse kaiken, mitä se tarvitsee tehtäviensä asianmukaiseen suorittamiseen. - Mitokondrioiden DNA: lla on hiukan erilainen geneettinen koodi kuin ydin -DNA: lla. Tämä johtaa useisiin eroihin proteiinien valmistuksessa: jos tietty nukleotidisekvenssi ydin -DNA: ssa johtaa tietyn proteiinin muodostumiseen, sama sekvenssi mitokondrio -DNA: ssa johtaa hieman erilaisen proteiinin muodostumiseen.
- Mitokondrio-DNA: ssa on hyvin vähän ei-koodaavia nukleotidisekvenssejä, eli ne eivät tuota proteiineja, tRNA: ita tai rRNA: ita. Prosentuaalisesti mitokondrioiden DNA: sta vain 3% on koodaamatonta.
Toisaalta ydin-DNA koodaa vain 7%, joten se sisältää paljon ei-koodaavia nukleotidisekvenssejä (jopa 93%).
Taulukko: yhteenveto ihmisen mitokondrioiden DNA: n ja ihmisen ydin -DNA: n välisistä eroista.
Mitokondrioiden DNA
Ydin -DNA
- Se on pyöreä
- Se on lineaarinen
- Siinä on yhteensä 16 569 typpipitoista emäsparia
- Siinä on yhteensä 3,3 miljardia typpipitoista emäsparia
- Se sisältää yhteensä 37 geeniä
- Se sisältää 20 000 - 25 000 geeniä
- Toimiakseen oikein se tarvitsee joidenkin ydin -DNA: sta peräisin olevien geenituotteiden tuen
- Se on itsenäinen ja tuottaa itse kaiken tarvittavan tehtäviensä suorittamiseksi
- Sitä voi esiintyä useissa kopioissa kussakin yksittäisessä mitokondrioissa
- Se on ainutlaatuinen, eli se on vain yksi kopio ja se sijaitsee ytimessä
- 97% sen muodostavasta nukleotidisekvenssistä on koodaavaa
- Vain 7% sen muodostavasta nukleotidisekvenssistä on koodaavaa
- Sitä ei ole järjestetty kromosomeiksi
- Se on jaettu 23 kromosomiin
- Se käyttää geneettistä koodia, joka on hieman erilainen kuin niin sanottu "perinteinen"
- Käytä "perinteistä" geneettistä koodia
- Sen perintö on äiti
- Sen perintö on puoliksi äiti ja puolet isä
- Jotkut sen nukleotidit osallistuvat kahteen geeniin samanaikaisesti
- Geenit muodostavat nukleotidisekvenssit erottuvat toisistaan hyvin
Perintö
Mitokondrioiden DNA -perintö on ehdottomasti äiti.
Tämä tarkoittaa sitä, että vanhempien parissa nainen lähettää mitokondrioiden DNA: n jälkeläisille (eli lapsille).
Täysin päinvastaisella tavalla kuin edellä, ydin -DNA -perintö on puoliksi äidin ja puoliksi isän eli toisin sanoen molemmat vanhemmat osallistuvat tasavertaisesti ydin -DNA: n siirtoon jälkeläisissä.
Huomaa: mitokondrioiden DNA: n perintöön äidiltä liittyy myös mitokondriorakenne. Näin ollen yksilössä olevat mitokondriot ovat äitiä.
Liittyvät patologiat
Lähtökohta: Geneettinen mutaatio on pysyvä muutos nukleotidisekvenssissä, jotka muodostavat ydin- tai mitokondrio -DNA -geenin.
Tyypillisesti geneettisen mutaation läsnäolo johtaa "geenin normaalin toiminnan muutokseen tai menetykseen.
Mutaatioiden esiintyminen mitokondrioiden DNA -geeneissä voi johtaa monenlaisiin sairauksiin, mukaan lukien:
- Leberin perinnöllinen optinen neuropatia
- Kearns-Sayren oireyhtymä
- Leighin oireyhtymä
- Sytokromi C -oksidaasin puute
- Progressiivinen ulkoinen oftalmoplegia
- Pearsonin oireyhtymä
- Mitokondrioiden enkefalomyopatia, johon liittyy maitohappoasidoosi ja aivohalvaus (MELAS-oireyhtymä)
- Diabetes, johon liittyy äidin kuurous
- Myokloninen epilepsia, jossa on epäsäännöllisiä punaisia kuituja
Mitä tulee patologisiin tiloihin, jotka liittyvät yhteen tai useampaan mitokondrioiden DNA -mutaatioon, kaksi näkökohtaa on selvennettävä.
Ensinnäkin taudin vakavuus riippuu mutatoituneiden mitokondrioiden DNA: iden ja terveiden, normaalien mitokondrioiden DNA: iden välisestä kvantitatiivisesta suhteesta. Jos mutatoituneiden mitokondrioiden DNA: iden lukumäärä on huomattavasti suurempi kuin terveiden DNA: iden, tuloksena oleva tila on vakavampi.
Toiseksi mitokondrio -DNA: n mutaatiot vaikuttavat vain joihinkin kehon kudoksiin, erityisesti niihin, jotka vaativat suuria määriä hapettavaa fosforylaatioprosessia aiheuttavaa ATP: tä. toiminto, jonka mitokondrio -DNA normaalisti suorittaa.
LEBERIN PERINNÖLLINEN OPTINEN NEUROPATIA
Leberin perinnöllinen optinen neuropatia syntyy peräti neljän mitokondrio -DNA -geenin mutaation seurauksena. Nämä geenit sisältävät tietoa, joka johtaa niin sanotun kompleksin I (tai NADH-oksidireduktaasin) synteesiin, joka on yksi erilaisista entsyymeistä, jotka osallistuvat oksidatiiviseen fosforylaatioprosessiin.
Patologian ilmenemismuotoja ovat näköhermon asteittainen rappeutuminen ja näön asteittainen menetys.
KEARNS-SAYRE-SYNDROMI
Kearns-Sayren oireyhtymä ilmenee johtuen siitä, että mitokondrioiden DNA: ta ei ole riittävästi (HUOM: tietyn nukleotidisekvenssin puuttumista kutsutaan deleetioksi).
Ihmiset, joilla on Kearns-Sayren oireyhtymä, kehittävät oftalmoplegiaa (silmän ja lihasten täydellinen tai osittainen halvaus), retinopatian ja sydämen rytmihäiriöiden muotoa (eteis-kammiokatkos).
LEIGHIN SYNDROMI
Leighin oireyhtymä johtuu mitokondrioiden DNA-mutaatioista, jotka voivat vaikuttaa ATP-syntaasiproteiiniin (jota kutsutaan myös V-kompleksiksi) ja / tai joihinkin tRNA: iin.
Leighin oireyhtymä on etenevä neurologinen sairaus, joka ilmenee lapsuudessa tai lapsuudessa ja joka on vastuussa kehityksen viivästymisestä, lihasheikkoudesta, perifeerisestä neuropatiasta, motorisista häiriöistä, hengitysvaikeuksista ja oftalmoplegiasta.
SYTOKROMI C -OKSIDAASIN VIKA
Sytokromi C -oksidaasin puute johtuu vähintään kolmen mitokondrio -DNA -geenin mutaatiosta. Nämä geenit ovat välttämättömiä sytokromi C -oksidaasi (tai kompleksi IV) entsyymin, joka osallistuu hapettavaan fosforylaatioprosessiin, oikean synteesin kannalta.
Tyypillisiä sytokromi C -oksidaasin puutteen ilmenemismuotoja ovat: luustolihasten toimintahäiriö, sydämen vajaatoiminta, munuaisten vajaatoiminta ja maksan vajaatoiminta.
EDISTYVÄ ULKOINEN OPTALMOPLEGIA
Progressiivinen ulkoinen oftalmoplegia johtuu huomattavan määrän mitokondrioiden DNA -nukleotidien puutteesta (poistettu)
Tämä patologia, jolla on progressiivinen luonne (kuten nimestä voi arvata), aiheuttaa silmämotoristen lihasten halvaantumisen, mistä seuraa ptoosi ja huomattavia näköongelmia.
PEARSONIN SYNDROOMI
Pearsonin oireyhtymä ilmenee mitokondrioiden DNA: n näkyvän poistamisen jälkeen, samalla tavalla kuin etenevä ulkoinen oftalmoplegia ja Kearns-Sayren oireyhtymä.
Pearsonin oireyhtymän tyypillisiä ilmenemismuotoja ovat: sideroblastinen anemia, haiman toimintahäiriö (esim. Insuliiniriippuvainen diabetes), neurologiset vajaatoiminnot ja lihashäiriöt.
Pearsonin oireyhtymä yleensä aiheuttaa sairastuneen kuoleman nuorena. Itse asiassa tämän patologian kärsineet saavuttavat harvoin aikuisuuden.
MELAS -SYNDROMI
MELAS-oireyhtymä, joka tunnetaan myös nimellä mitokondrioiden enkefalomyopatia, johon liittyy maitohappoasidoosia ja aivohalvauksen kaltaisia jaksoja, johtuu vähintään 5 mitokondrio-DNA-geenin mutaatiosta.
Nämä geenit edistävät NADH-oksidireduktaasin tai kompleksin I ja joidenkin tRNA: iden synteesiä.
MELAS -oireyhtymään liittyy neurologisia häiriöitä, lihassairauksia, maitohapon epätavallista kertymistä kudoksiin (ja kaikki siihen liittyvät oireet), hengitysvaikeuksia, suolen toiminnan heikkenemistä, toistuvaa väsymystä, munuaisongelmia, sydänvaivoja, diabetesta, epilepsiaa ja koordinaation puute.
MUUT PATOLOGIAT
Erilaisten tieteellisten tutkimusten mukaan sairaudet, kuten syklisen oksentelun oireyhtymä, retinitis pigmentosa, ataksia, Parkinsonin tauti ja Alzheimerin tauti, näkisivät myös mitokondrioiden DNA: n ja joidenkin sen mutaatioiden osallistumisen.