Määritelmä talassemia
Thalassemia on geneettisesti tarttuva verisairaus, jossa keho syntetisoi epänormaalin hemoglobiinin muodon.
Kuten useimmat ihmiset tietävät, hemoglobiini on punasolujen sisältämä proteiini, joka on välttämätön hapen kuljetukselle veressä. Talassemiaa sairastavilla hemoglobiinin mutatoitu muoto aiheuttaa asteittaisen mutta väistämättömän punasolujen tuhoutumisen anemiaan asti.
Lääketieteellisten tilastojen perusteella on selvää, että talassemia vaikuttaa lähinnä Lähi -idän maiden, Afrikan maiden ja kaikkien suoalueiden asukkaisiin (ei ole yllättävää, että talassemiaa kutsutaan myös Välimeren anemia).
Luokittelu ja syyt
Viallisen proteiini -alayksikön (joka muodostaa hemoglobiinin) mukaan erotetaan kaksi talassemian muotoa; ennen kuin aloitamme analyysin, otamme askeleen taaksepäin selventämään joitakin erittäin tärkeitä käsitteitä.
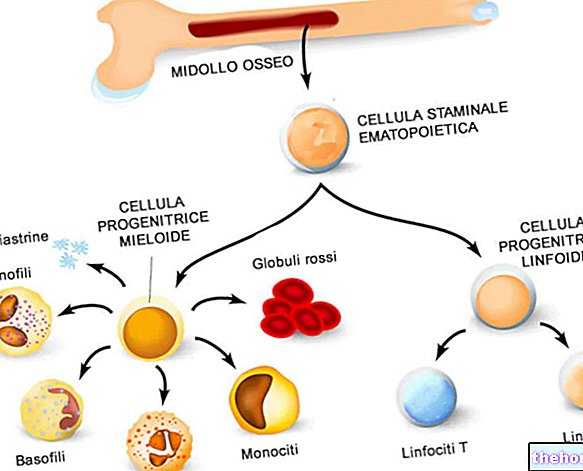
Hemoglobiini on par excellence -kantaja, jota käytetään hapen kuljettamiseen veressä; se koostuu kahdesta proteiinista, jotka tunnetaan alfa-globuliinina ja beeta-globuliinina.
Thalassemia ilmenee, kun yksi tai useampi geeni, jotka ohjaavat yhden tai molempien näiden proteiinien tuotantoa, on viallinen (mutatoitunut).
Thalassemia johtuu hemoglobiinin muodostavien proteiinien DNA -mutaatiosta: nämä muutokset vaikuttavat voimakkaasti hemoglobiinin fysiologiseen synteesiin ja johtavat anemiaan tuhoamalla punasolut.
Talassemian luokittelu on tehtävä kahden tärkeän tekijän perusteella:
- Vanhemmilta perittyjen mutatoituneiden geenien lukumäärä
- Mukana olevan proteiinin tyyppi (alfa- tai beeta -hemoglobiini)
Alfa -talassemia
Talassemian "alfa" -muodossa - jossa hemoglobiinin 4 "alfa" pallomaista alayksikköä (kromosomissa 16) voidaan mutatoida - on mukana yksi tai useampi viallinen geeni; jokainen pallomainen alayksikkö on selvästi koodattu geenistä, joten geenit, joihin voi osallistua, ovat 4.

Yleinen oirekuva muuttuu vakavammaksi, kun mukana on kolme tai neljä geeniä: ensimmäisessä tapauksessa puhumme "hemoglobiini H -tauti"(Kohtalaisia tai vakavia oireita). Kun kaikki neljä geeniä ovat mukana, tautia kutsutaan suuri alfa-talassemia: vastaavissa tilanteissa vastasyntynyt kuolee juuri ennen syntymää tai pian sen jälkeen.
Beeta -talassemia
Talassemian beeta -muoto, kuten voidaan arvata, tapahtuu, kun beeta -ketjujen koostumukseen osallistuvat geenit ovat mutaatioita (kromosomin 11 tasolla): tässä tapauksessa vain kaksi geeniä voi vaikuttaa. Jos vain yksi geeni muuttuu, sitä kutsutaan beeta-talassemia vähäinen, jossa potilas valittaa oleellisista oireista. Samoin kuin alfa -variantti, molempien geenien osallistuminen hemoglobiinin beeta -ketjuihin johtaa yhteen suuri beeta-talassemia (tai Cooleyn anemia), joka kuvastaa vakavia ja vakavia oireita; tässä tapauksessa oireet alkavat kuitenkin yleensä parin vuoden kuluttua syntymästä.
Katso video
- Katso video youtubesta
Oireet
Lisätietoja: Thalassemian oireet
Thalassemia on erittäin vakava perinnöllinen sairaus, niin paljon, että jotkut sen muunnelmista, kuten alfa-thalassemia major, voivat aiheuttaa vauvan kuoleman synnytyksen aikana tai pian syntymän jälkeen. ensimmäiset oireet parin vuoden kuluessa syntymästä (vaikea anemia).
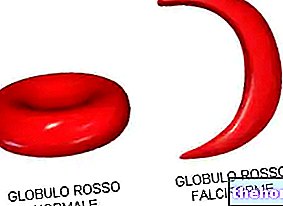
Jos vain yksi geeni muuttuu sekä talassemian alfa- että beeta -muodossa, potilaat eivät valittaa havaittavista oireista; vain potilaalta otetun verinäytteen mikroskooppianalyysin avulla on havaittu poikkeavuuksia punasolujen muodossa ja rakenteessa, paljon normaalia pienempiä.
Anemian lisäksi talassemiapotilailla voi esiintyä yksi tai useampi seuraavista oireista: väsymys, mielialan muutokset (ärtyneisyys), kasvun vajaatoiminta, kasvojen luun epämuodostumat, keltaisuus, hengenahdistus ja tumma virtsa.
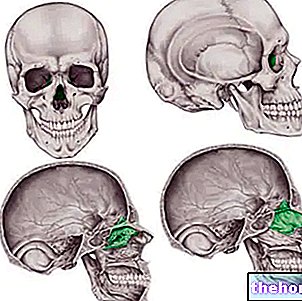
Vakavuustapauksissa oireinen kuva talassemiapotilaasta voi rappeutua ja aiheuttaa todellisia luun epämuodostumia, erityisesti kasvoissa ja kallossa; talassemia voi edistää "luuytimen epänormaalia laajentumista sekä tekemällä luumassasta hauraan että lisäämällä valtavasti luunmurtumien riskiä.
Talassemian komplikaatioista on mainittava myös mahdollinen raudan kertyminen (hemokromatoosi), joka ilmentää sekä itse sairautta että potilaan tarvitsemia toistuvia verensiirtoja.
Thalassemia aiheuttaa usein splenomegaliaa, eli pernan liiallista volumetristä nousua: tämä patologinen kliininen tila vaatii usein pernan poistoa, elimen kirurgista poistamista. Kuten tiedämme, perna on tärkeä elin, jota käytetään verisolujen synteesiin ja vasta -aineet infektioiden hallinnan lisäksi: sen poistaminen suosii selvästi puolustusfunktion heikkenemistä bakteeri- ja virushyökkäyksiä vastaan, mikä tekee potilaasta herkemmän infektioille. On kuitenkin huomattava, että myös talassemia lisää riskiä tartunnan saamiseksi: jos perna poistetaan talassemian yhteydessä, tartuntamahdollisuudet lisääntyvät liioitellusti.

Diagnoosi
Jos isä ja / tai äiti kärsii talassemiasta, todennäköisyys tartuttaa tauti jälkeläisille on erittäin suuri. Olemme analysoineet, että kaikki talassemian muodot eivät alkaneet täsmällisellä oireella heti syntymästään lähtien: vastaavissa tilanteissa, jos epäillään talassemiaa, potilaalle voidaan suorittaa useita erityisiä testejä ja tutkimuksia, joiden tarkoituksena on diagnoosi ( kuten hemoglobiini A2: n määrittäminen, joka on koholla terveillä tutkittavilla, joilla on beeta-talassemisia geenejä).
Fysikaalisten tutkimusten joukossa pernan lääketieteellinen tunnistus voi joskus todeta talassemian: splenomegalia, kuten aiemmin mainittiin, on ensimmäinen hälytyssignaali Välimeren anemiasta. Verikokeet ovat tarkempia ja tarkempia: talassemian verinäytteessä punasolut näyttävät mikroskoopilla katsottuna pieniltä ja muodoltaan epänormaaleilta. Lisäksi Thalassemiaa sairastavan potilaan huolellinen verenkuva paljastaa vakavan anemian: tämä testi on hyödyllinen raudan määrän laskemisessa veressä, DNA -analyysin suorittamisessa taudin diagnosoimiseksi ja hemoglobiinin mahdollisen mutaation arvioimiseksi .
Toisaalta hemoglobiinien elektroforeesi paljastaa happea kuljettavien proteiinien epänormaalin muodon.
Joitakin talassemian variantteja ei voida diagnosoida elektroforeesilla: tässä tapauksessa potilaalle tehdään "mutaatioanalyysitesti", joka on hyödyllinen talassemian havaitsemiseksi ja todentamiseksi.
Lääkkeet ja hoidot
Katso myös: Lääkkeet talassemian hoitoon
Koska kyseessä on geneettisesti tarttuva tauti, on ymmärrettävää, että tällä hetkellä ei ole lääkettä, joka kykenisi kääntämään taudin; On kuitenkin mahdollista hallita oireita ja parantaa potilaan elämänlaatua. Yhden hoidon valinta toisen sijaan riippuu talassemian tyypistä ja oireiden vakavuudesta.
Talassemian lievässä muunnoksessa (jossa esimerkiksi vain yksi geeni muuttuu) lääkkeitä ei tarvita, koska potilas ei valita oireista. Tällaisissa olosuhteissa on silti suositeltavaa suorittaa tarvittavat tarkastukset säännöllisesti; Joskus verensiirrot ovat joskus hyödyllisiä (etenkin leikkauksen ja synnytyksen tapauksessa).
Keskivaikeissa tai vaikeissa oireisissa muodoissa hoitomenetelmä on erilainen ja voi vaatia usein verensiirtoja tai vakavissa tapauksissa kantasolusiirtoa.
- Verensiirrot: tämä terapeuttinen lähestymistapa voi myös aiheuttaa vakavia komplikaatioita, koska toistuvat verensiirrot voivat edistää raudan patologista kertymistä vereen (hemokromatoosi), joka vaatii erityishoitoa, jolla pyritään poistamaan raudan varastointi, joka tunnetaan terapiakelaattorina (lääkkeillä, kuten Deferasirox ja Deferiproni).Lisätietoja: lue artikkeli lääkkeistä hemokromatoosin hoitoon.
- Luuydinsiirto: varattu vakavimmille tapauksille, joissa talassemia aiheuttaa vakavia toimintahäiriöitä kehossa.