Yleisyys
Retinoblastooma (Rb) on pahanlaatuinen silmän kasvain, joka kehittyy verkkokalvon soluista. Tämä syöpä voi esiintyä missä iässä tahansa, mutta puhkeaminen on yleisintä ennen 5 -vuotiaita.

Lapsuuden syöpä on aggressiivinen: Retinoblastooma voi levitä imusolmukkeisiin, luisiin tai luuytimeen. Harvoin se liittyy keskushermostoon (aivot ja selkäydin).
Noin 90 prosentilla retinoblastoomaa sairastavista lapsista on positiivinen ennuste (parantumisen todennäköisyys), jos diagnoosi on varhainen ja hoito aloitetaan ennen kuin syöpä leviää. Aina kun mahdollista, lääketieteellisen toimenpiteen tavoitteena on säilyttää potilaan näkö.
Syyt
Kasvaimen puhkeamiseen johtava tapahtumasarja on monimutkainen.Tämä alkaa, kun verkkokalvon solut kehittävät mutaation (tai deleetion), johon liittyy RB1 -tuumorisuppressorigeeni, joka sijaitsee kromosomin 13 (13q14) q14 -kaistalla.
Jokaisessa solussa on normaalisti kaksi RB1 -geeniä:
- Jos ainakin yksi geenikopio toimii oikein, retinoblastoomaa ei synny (mutta riski kasvaa);
- Kun molemmat geenikopiot ovat mutatoituneet tai puuttuvat, tapahtuu hallitsematonta solujen lisääntymistä.
Monissa tapauksissa on epäselvää, mikä aiheuttaa muutoksia RB1 -geenissä (satunnainen retinoblastooma); nämä voivat johtua satunnaisista geneettisistä virheistä, joita esiintyy esimerkiksi lisääntymisen ja solujakautumisen aikana. On kuitenkin tiedossa, että retinoblastooman taustalla olevat geneettiset poikkeavuudet voidaan siirtää myös vanhemmilta lapsille, joilla on autosomaalinen hallitseva perintömalli. Tämä tarkoittaa, että jos vanhempi kantaa mutatoitunutta (hallitsevaa) geeniä, jokaisella lapsella on 50% mahdollisuus periä se ja 50% todennäköisyys saada normaali geneettinen rakenne (resessiiviset geenit).
- Satunnainen solu inaktivoi ainoan normaalin kopionsa RB1 -geenistä (yksi kopio on jo mutatoitunut);
- Kahden RB1 -kopion menetys johtaa "verkkokalvon liialliseen lisääntymiseen.
- Satunnainen solu inaktivoi yhden normaaleista RB1 -geeneistään;
- RB1 -geenin toinen kopio on inaktivoitu;
- Kahden RB1 -kopion menetys aiheuttaa liiallisen soluproliferaation, mikä johtaa retinoblastoomaan.
Geneettiset ja molekyyliominaisuudet
- Retinoblastooma oli ensimmäinen kasvain, joka liittyi suoraan "geneettiseen poikkeavuuteen (kromosomin 13 q14 -kaistan deleetio tai mutaatio).
- RB1 koodaa pRb -proteiinia, jolla on keskeinen rooli solusyklissä: se mahdollistaa DNA: n replikaation ja solusyklin etenemisen, koska se osallistuu S -vaiheen geenien transkription säätelyyn (G1 → † "S).
- Retinoblastooman lisäksi RB1 -geeni inaktivoituu virtsarakon, rintojen ja keuhkosyövissä.
Perinnöllinen retinoblastooma
Lapsilla, joilla on perinnöllinen retinoblastooma, on taipumus kehittää tauti aikaisemmassa iässä kuin satunnaisia tapauksia. Lisäksi näillä lapsilla on suurempi riski saada muita ei-silmäsyöpiä, koska RB1-geenin poikkeavuus on synnynnäinen (eli läsnä syntymästä lähtien) ja vaikuttaa kaikkiin kehon soluihin (tunnetaan ituradan mutaationa), myös molempien solut. verkkokalvot: Tästä syystä lapsilla, joilla on perinnöllinen muoto, on usein kahdenvälinen retinoblastooma eikä vain yksi silmä.
Oireet
Lisätietoja: Retinoblastooman oireet
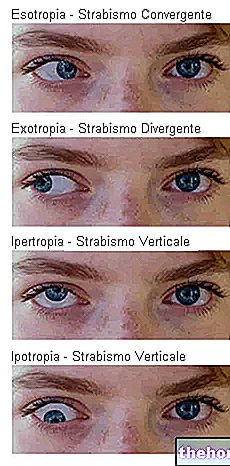
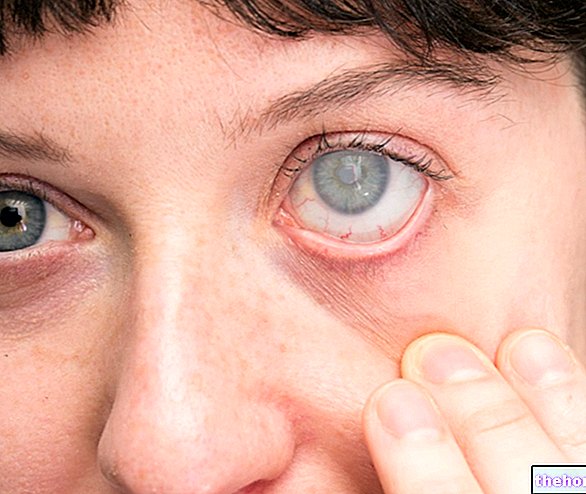
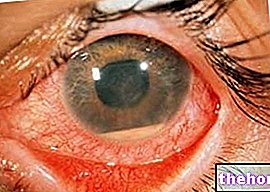
Yleisin ja ilmeisin merkki retinoblastoomasta on pupillin epänormaali ulkonäkö, joka antaa harmahtavan valkoisen heijastuksen, kun siihen osuu valonsäde (leukokoria tai amauroottinen kissan refleksi)). Muita merkkejä ja oireita ovat: heikentynyt näkö, silmäkipu ja punoitus sekä kehityshäiriö. Joillekin retinoblastoomaa sairastaville lapsille voi kehittyä silmänräpäys (väärin kohdistetut silmät); muissa tapauksissa on mahdollista löytää neovaskulaarinen glaukooma, joka jonkin ajan kuluttua voi aiheuttaa silmän laajentumisen (buftalmo).
Syöpäsolut voivat edelleen tunkeutua silmään ja muihin rakenteisiin:
- Silmänsisäinen retinoblastooma. Retinoblastooma voidaan määritellä silmänsisäiseksi, kun kasvain sijaitsee kokonaan silmän sisällä. Kasvain löytyy vain verkkokalvosta tai vaikuttaa myös muihin osiin, kuten koroidiin, silmärungoon ja näköhermon osaan. Siksi silmänsisäinen retinoblastooma ei leviä silmän ulkopuolisiin kudoksiin.
- Extraokulaarinen retinoblastooma.Kasvain voi lisääntyä ja vaikuttaa silmän ympärillä oleviin kudoksiin (orbitaalinen retinoblastooma). Syöpä voi levitä myös muille kehon alueille, kuten aivoihin, selkärankaan, luuytimeen ja imusolmukkeisiin (metastaattinen retinoblastooma).
Kiertoradan laajentuminen, uveaalinen osallistuminen ja näköhermon hyökkäys ovat tunnettuja riskitekijöitä metastaattisen retinoblastooman kehittymiselle.
Diagnoosi
Jos perhehistoria on positiivinen, potilas käy säännöllisesti silmätutkimuksissa syöpäseulontaa varten. Jos synnynnäinen retinoblastooma on kahdenvälinen, se diagnosoidaan yleensä ensimmäisenä elinvuotena, kun taas kun se vaikuttaa vain yhteen silmään, kasvaimen läsnäolo voidaan vahvistaa noin 18-30 kuukauden iässä.
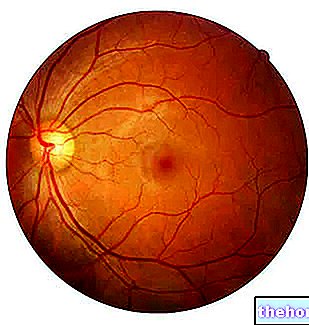
Retinoblastooman kliininen diagnoosi vahvistetaan silmänpohjan tutkimuksella, ja kasvain voi sijainnista riippuen olla näkyvissä yksinkertaisen silmätutkimuksen aikana epäsuoran oftalmoskopian avulla. Kuvantamistekniikoilla voidaan vahvistaa diagnoosi, määrittää kasvaimen vaihe (missä se on, kuinka laaja se on, vaikuttaako se muiden elinten toimintoihin jne.) Ja määrittää, onko hoito ollut tehokasta . Tutkimuksia voivat olla ultraääni, tietokonetomografia (CT) ja magneettikuvaus (MRI).
Molekyyli-geneettinen diagnoosi on mahdollista tunnistamalla RB1-geenin mutaatio. Perifeerisen veren lymfosyyttien sytogeneettistä analyysiä (eli kromosomien) käytetään kromosomin 13 (13q14.1-q14.2) deleetioiden tai uudelleenjärjestelyjen havaitsemiseen. .
Hoidot
Retinoblastooman tapauksessa voidaan käyttää useita hoitovaihtoehtoja.
Hoidon tavoitteet ovat:
- Poista kasvain ja pelasta potilaan henki;
- Säästä silmä jos mahdollista;
- Säilytä visio mahdollisimman paljon;
- Vältä muiden syöpien kehittymistä, jotka voivat myös johtua hoidosta, etenkin lapsilla, joilla on perinnöllinen retinoblastooma.
Ennuste (toipumisen todennäköisyys) ja hoitovaihtoehdot riippuvat seuraavista tekijöistä:
- Kasvaimen vaihe;
- Potilaan ikä ja yleinen terveydentila;
- Tuumoripesien sijainti, koko ja määrä;
- Syövän leviäminen muille alueille silmämunan lisäksi
- Kuinka todennäköistä on, että näkö voidaan säilyttää yhdessä tai molemmissa silmissä.
Useimmat retinoblastooman tapaukset diagnosoidaan varhaisessa vaiheessa ja hoidetaan onnistuneesti, ennen kuin syöpä voi etäpesäkkeitä silmämunan ulkopuolelle, mikä johtaa yli 90%: n paranemiseen.