Yleisyys
Osteogenesis imperfecta on synnynnäinen geneettinen sairaus, joka ei liity sukupuoleen ja joka on vastuussa tietystä luun hauraudesta ja huomattavasta taipumuksesta murtumiin.
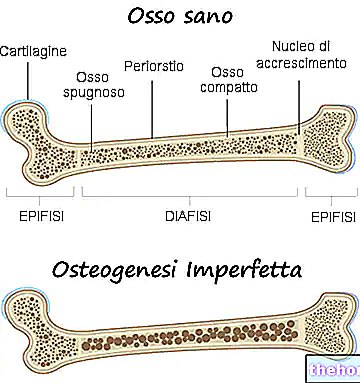
Osteogeneesin epätäydellisyyden oireita on lukuisia; yleensä ne koostuvat seuraavista: luun heikkeneminen, suuri taipumus luunmurtumiin, sinisten, harmaiden tai violettien silmäkalvon esiintyminen, luun epämuodostumien tai muiden luuston muutosten esiintyminen, kolmiomaiset kasvot, hampaiden hauraus jne. .
Yleensä seuraavat ovat välttämättömiä osteogenesis imperfectan oikean diagnoosin tekemiseksi: fyysinen tarkastus, sairaushistoria, lääketieteelliset kuvantamistestit, tyypin I kollageenin arviointitesti ja geneettinen testi.
Valitettavasti tällä hetkellä ainoat hoidot, jotka ovat saatavilla potilaille, joilla on osteogenesis imperfecta, ovat oireisia. Kyseinen sairaus on itse asiassa parantumaton.
Mikä on osteogenesis imperfecta?
Osteogenesis imperfecta on geneettinen sairaus, joka tekee potilaan luista heikompia ja alttiimpia murtumille.
Todellisuudessa termillä osteogenesis imperfecta lääkärit viittaavat heterogeeniseen geneettisten sairauksien ryhmään, jolle on ominaista tietty luun hauraus. Siksi on olemassa useita osteogenesis imperfectan muotoja (tai tyyppejä), jotkut paljon vakavampia kuin toiset.
Se on synnynnäinen sairaus
Ihmisillä, joihin se vaikuttaa, osteogenesis imperfecta on sairaus, joka on läsnä syntymästä lähtien, joten se voidaan määritellä kaikilta osin synnynnäiseksi sairaudeksi.
ONKO SEKSI SEURANTAISTA?
Osteogenesis imperfecta ei ole sukupuoleen liittyvä geneettinen sairaus, kuten hemofilia tai Klinefelterin oireyhtymä.
EPIDEMILOGIA
Joidenkin tilastotutkimusten mukaan osteogenesis imperfectan esiintyvyys olisi yhtä tapausta joka 15 000-20 000 syntymää kohden. Tämä tarkoittaa, että jokaisella 15 000–20 000 vastasyntyneellä on osteogenesis imperfecta.
Muut tilastolliset tutkimukset ovat myös osoittaneet, että osteogenesis imperfecta vaikuttaa miehiin ja naisiin tasapuolisesti eikä sillä ole etusijaa tiettyä väestöä tai etnistä ryhmää kohtaan.
Elinikä on erittäin vaihteleva parametri, joka riippuu osteogenesis imperfectan muodosta.
Syyt
Osteogenesis imperfecta johtuu lähes aina tyypin I kollageenin tuotannon laadullisesta ja määrällisestä muutoksesta.
Tyypin I kollageeni on välttämätön luiden vahvistamiseksi ja terveiden sidekudosten, kuten ruston, jänteiden, ihon, silmän kalvon jne., Ylläpitämiseksi.
Siksi tyypin I kollageenin tuotannon muutos vaikuttaa luuston lujuuteen ja ihmiskehossa olevien sidekudosten terveyteen.
Mikä muuttaa kollageenin tuotantoa?
Geneettinen sairaus on tila, joka johtuu yhden tai useamman solun DNA: ta muodostavan geenin mutaatiosta.
Osteogenesis imperfectan tapauksessa jälkimmäisen syyt löytyvät lähes aina yhden tai molempien geenien COL1A1 (sijaitsee kromosomissa 17) ja COL1A2 (sijaitsee kromosomissa 7) mutaatiosta.
Normaaleissa olosuhteissa COL1A1 ja COL1A2 säätelevät tyypin I kollageenin normaalia tuotantoa; kun varauksessa on mutaatioita, he epäonnistuvat säätelytoiminnassaan.
Tärkeää: mitkä muut geenit, jos ne ovat mutatoituneita, aiheuttavat osteogenesis imperfectaa?
COL1A1- ja COL1A2 -mutaatioiden lisäksi IFITM5-, SERPINF1-, CRTAP- ja LEPRE1 -geenien mutaatiot ovat mahdollisia osteogenesis imperfectan syitä.
Edellä mainitut geenit kattavat eri toiminnot kuin COL1A1 ja COL1A2 - siksi ne eivät kontrolloi tyypin I kollageenin tuotantoa - mutta niillä on silti "vaikutus ihmisen luuston luuston lujuuteen ja vastustuskykyyn".
MITÄ GENEETTISTÄ TAUDISTA SE ON?
Osteogenesis imperfecta on autosomaalinen geneettinen sairaus.
Termi autosomaalinen, johon liittyy geneettinen sairaus, osoittaa, että kyseinen tila johtuu geneettisistä mutaatioista, jotka perustuvat autosomaalisiin ja ei-sukupuolisiin kromosomeihin.
Lukijoita muistutetaan, että ihmisellä on kromosomijoukko, jossa on 23 paria kromosomeja, joista 22 paria on autosomaalista tyyppiä ja vain yksi pari on seksuaalista tyyppiä. yksilö.
COL1A1-, COL1A2- ja IFITM5 -mutaatioiden jälkeisellä osteogenesis imperfectalla on kaikki autosomaalisen hallitsevan sairauden ominaisuudet.Se johtuu geenien SERPINF1, CRTAP ja LEPRE1 mutaatioista ja sillä on autosomaalisen resessiivisen taudin piirteitä.
TYYPIT
Tällä hetkellä lääkärit uskovat, että osteogenesis imperfectaa on 8 tyyppiä (tai muotoa). Eri tyyppien erottamiseksi he päättivät käyttää roomalaista numerointia, tarkemmin ottaen kahdeksan ensimmäistä roomalaista numeroa.
Seuraavassa taulukossa esitetään 8 osteogenesis imperfecta -muotoa, niitä aiheuttavat mutaatiot ja muut ominaisuudet.
Kaveri
Mutoitunut geeni
Geneettisen sairauden tyyppi
THE
COL1A1
Autosomaalinen hallitseva
II
COL1A1 ja COL1A2
Autosomaalinen hallitseva
III
COL1A1 ja COL1A2
Autosomaalinen hallitseva
IV
COL1A1 ja COL1A2
Autosomaalinen hallitseva
V.
IFITM5
Autosomaalinen hallitseva
SINÄ
SERPINF1
Autosomaalinen resessiivinen
VII
CRTAP
Autosomaalinen resessiivinen
VIII
HARE 1
Autosomaalinen resessiivinen
* HUOM: tietysti COL1A1- ja COL1A2 -mutaatiot, jotka aiheuttavat neljä ensimmäistä osteogenesis imperfecta -muotoa, ovat geneettisiä muutoksia, joilla on hieman erilaiset ominaisuudet. Muuten ei olisi mitään järkeä erottaa toisiaan muista.
Oireet, merkit ja komplikaatiot
Kaikentyyppiset osteogenesis imperfecta ovat vastuussa luiden heikkenemisestä siten, että taudista kärsivällä henkilöllä on erityinen alttius murtumille. Luiden heikkenemisaste vaihtelee muodon mukaan; joillekin näistä heikkeneminen on suurempi kuin toisille.
Tämän sanottuaan on korostettava, että jokaisella osteogenesis imperfecta -muodolla on oma oireinen kuva, joka joillekin saattaa muistuttaa muiden muotojen oireenmukaista kuvaa.
MAHDOLLISET Oireet ja merkit
Mahdollisia osteogenesis imperfectan oireita ja merkkejä ovat:
- Luun epämuodostumat;
- Lyhyt ja pieni runko (tarkoitettu runkoksi);
- Nivelongelmat (esim. Löysät nivelet);
- Lihas heikkous;
- Sininen, violetti tai harmaa silmän sklera;
- Kolmion muotoiset kasvot;
- Tynnyri rinnassa;
- Selkärangan morfologiset poikkeavuudet;
- Hampaiden hauraus;
- Kuulon heikkeneminen tai täydellinen menetys;
- Hengitysongelmia
- Ongelmia, jotka liittyvät tyypin 1 kollageenin puuttumiseen tai puutteeseen.
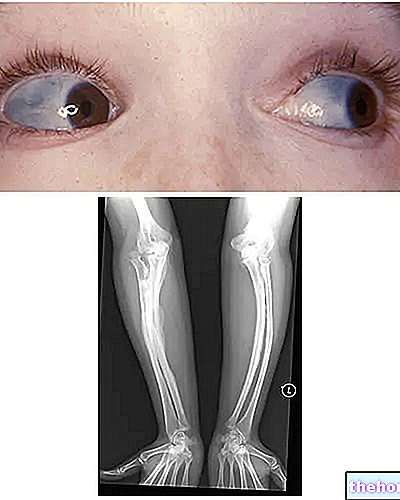
Osteogenesis Imperfecta: huomioi kaularangan sininen väri ja taudille ominaiset luun muodonmuutokset. Lähde: wikipedia.org
MITÄ OLEVAT VAKAVIMMAT MUODOT Epätäydellinen OSTEOGENEESI?
Lääkärit luokittelevat erilaisten osteogenesis imperfecta -tyyppien oireiden vakavuuden asteikolla 3 astetta, jotka ovat: lievä aste, kohtalainen aste ja vaikea aste.
Vain yksi muoto kuuluu "lievän asteen" luokkaan: "tyypin I osteogenesis imperfecta"; 4 osteogenesis imperfectan muotoa kuuluvat "kohtalaisen asteen" luokkaan: IV, V ja VI; lopuksi "vakavan asteen" luokkaan kuuluvat 3 lomakkeet: II, III, VII ja VIII.
TYYPPI I: OMINAISUUDET
Kaikista yleisimmällä ja vähiten vakavalla tyypin I osteogenesis imperfectalla on seuraavat ominaisuudet:
- Se aiheuttaa murtumia erityisesti ennen murrosikää;
- Sillä ei ole "juurikaan vaikutusta pituuteen, joten potilaat ovat yleensä normaalikorkeita";
- Aiheuttaa nivelongelmia ja lihasheikkoutta
- Se vastaa sinisestä, violetista tai harmaasta sklerasta;
- Se on kolmiomaisten kasvojen ja selkärangan poikkeavuuksien syy;
- Se ei melkein koskaan aiheuta luun epämuodostumia. Jos se provosoi heitä, ne ovat vähäisiä;
- Se voi aiheuttaa hampaiden haurautta ja / tai kuulon heikkenemistä (jälkimmäinen esiintyy yleensä aikuisiässä);
- Se liittyy tyypin I kollageeniin, joka on laadultaan normaalia, mutta määrältään epänormaalia (se on normaalia huonompi).
TYYPPI II: OMINAISUUDET
Tyypin II osteogenesis imperfectalle on ominaista:
- Kuolinsyy syntyessä tai pian sen jälkeen. Hengitysongelmat aiheuttavat lähes aina kuoleman;
- Huomattava luun hauraus ja vakavat luun epämuodostumat;
- Lyhytkasvuinen ja alikehittynyt keuhko
- Sininen, violetti tai harmaa sklera;
- Tyypin I kollageenin määrällisten ja laadullisten poikkeavuuksien esiintyminen.
TYYPPI III: OMINAISUUDET
Tyypin III osteogenesis imperfectalla on seuraavat ominaisuudet:
- Vaikka se on erittäin vakava, se ei useinkaan aiheuta kuolemaa vastasyntyneen aikana;
- Se liittyy "korkeaan luun haurauteen;
- Se on vastuussa lyhyestä kasvusta, nivelongelmista, lihasheikkoudesta (erityisesti jaloissa ja käsivarsissa), tynnyrin rinnasta, kolmiomaisista kasvoista ja selkärangan epänormaalista kaarevuudesta;
- Se johtuu sinisestä, violetista tai harmaasta sklerasta;
- Se voi aiheuttaa hengitysvaikeuksia, hampaiden haurautta ja kuulon heikkenemistä;
- Se on usein vastuussa luun epämuodostumista;
- Se liittyy tyypin I kollageenin laadullisiin ja määrällisiin poikkeavuuksiin.
TYYPPI IV: OMINAISUUDET
Tyypin IV osteogeneesille on tunnusomaista:
- Luun haurausaste muotojen II ja III ja muodon I välillä;
- Keskimääräistä lyhyempi kasvu;
- Sininen, violetti tai harmaa sklera;
- Lievän / kohtalaisen luun epämuodostumat, selkärangan ja tynnyrin rinnan vähäiset poikkeavuudet;
- Kolmion muotoiset kasvot;
- Mahdollinen hampaiden hauraus ja kuulon heikkeneminen;
- Tyypin I kollageenipoikkeavuuksia.
TYYPPI V: OMINAISUUDET
Tyypin V osteogenesis imperfecta muistuttaa jollain tavalla tyypin IV osteogeneesis imperfectaa. Sillä on kuitenkin joitain erityispiirteitä, joita ovat:
- Normaali värillinen sklera;
- Hampaiden haurauden puuttuminen;
- Epänormaalien luukarvojen muodostuminen murtuneiden luiden paranemisprosessin aikana;
- Säteen ja kyynärpään välissä olevan interosseous -kalvon kalkkeutuminen. Tämä heikentää kyynärvarren liikkuvuutta.
TYYPPI VI: OMINAISUUDET
Myös tyypin VI osteogenesis imperfecta on samanlainen kuin muoto IV. Sen erottamiseksi jälkimmäisestä on joitain erityispiirteitä, mukaan lukien alkalisen fosfataasin korkea pitoisuus veressä ja joidenkin luiden kalojen selkärangan kaltaisten lamellien esiintyminen joissakin luissa.
TYYPPI VII: OMINAISUUDET
Oireellisesti tyypin VII osteogenesis imperfecta voi joissakin olosuhteissa muistuttaa tyyppiä IV ja muissa olosuhteissa tyypin II.
Tämän vakavan patologisen muodon erityispiirteitä ovat:
- Lyhytkasvuinen;
- Erittäin lyhyen olkaluun (käsivarren luun) ja reisiluun (reisiluun) läsnäolo;
- Usein esiintyvä lonkan epämuodostuma, joka tunnetaan nimellä coxa vara.
TYYPPI VIII: OMINAISUUDET
Tyypin VIII osteogenesis imperfecta muistuttaa hyvin muotoja II ja III.
Sen erikoisominaisuuksista erottuvat seuraavat: vakava kasvuvaje, luuston vaikea hypomineralisaatio ja prolyyli-3-hydroksylaasientsyymin puute (tai vähäinen läsnäolo).
Diagnoosi
Yleensä diagnostinen prosessi, johon potilaat, joilla epäillään osteogenesis imperfecta -muotoa, kohdistetaan, alkaa huolellisella fyysisellä tutkimuksella ja huolellisella sairaushistorialla; sitten se jatkuu, "analysoimalla potilaan sukututkimusta ja tekemällä sarjan diagnostisia kuvantamistestejä (röntgenkuvat, CT-skannaukset jne.); lopuksi se päättyy kvantitatiiviseen ja laadulliseen arviointiin tyypin I kollageenista ja geneettinen testi.
Nykyään on mahdollista diagnosoida osteogenesis imperfecta jopa synnytystä edeltävässä vaiheessa altistamalla raskaana oleva nainen ultraäänelle.
TAVOITTEEN TUTKIMUKSEN JA HISTORIAN TÄRKEYS
Osteogenesis imperfecta -lääketieteen asiantuntija pystyy hyvin usein diagnosoimaan edellä mainitun sairauden jopa fyysisen tarkastuksen ja anamneesin avulla. Tämä tarkoittaa, että nämä diagnostiset testit eivät ole vähäpätöisiä.
I -tyypin kollageenituotannon arviointi
Tyypin I kollageenin laadullinen ja määrällinen arviointi on pääsääntöisesti erittäin luotettava testi, koska kuten todettiin, useimmille osteogenesis imperfecta -tapauksille on ominaista mutaatiot geeneissä, jotka ohjaavat tyypin 1 kollageenin tuotantoa.
Arvioidakseen yksilön solutasolla olevan tyypin I kollageenin määrää ja laatua lääkärit voivat luottaa ihon biopsiaan tai tiettyyn verikokeeseen.
Molemmat arviointitestit ovat melko monimutkaisia ja potilas (tai hänen vanhempansa) saattaa joutua odottamaan useita viikkoja saadakseen tietää tulokset.
GENEETTINEN TESTI
Geenitestin avulla, joka koettelee tutkittavan yksilön koko DNA: ta, lääkärit voivat päättää lopullisesti esiintyvän geneettisen mutaation ominaisuuksista.
Yleensä geneettisen testin suorittamista kaikelle solu -DNA: lle suunnitellaan, kun tyypin I kollageenin ominaisuuksien arviointi ei ole tuottanut toivottuja tuloksia tai kun COL1A1- tai COL1A2 -mutaatio ei aiheuta "osteogenesis imperfecta".
PRENATAALINEN DIAGNOOSI
Prenataalinen ultraääni on erittäin hyödyllinen tyypin II ja tyypin III osteogenesis imperfectan tunnistamisessa.
Hoito
Tällä hetkellä ei ole olemassa erityistä parannuskeinoa osteogenesis imperfectalle. Toisin sanoen osteogenesis imperfecta -potilaiden on määrä elää edellä mainitun tilan kanssa kuolemaan saakka, mikä johtuu usein itse taudin seurauksista.
Spesifisen hoidon puute ei sulje pois muiden hoitomuotojen olemassaoloa. Itse asiassa osteogenesis imperfecta -potilaan hoitomahdollisuuksiin kuuluu erilaisia oireenmukaisia hoitoja; oireenmukaisilla hoidoilla tarkoitetaan hoitoja, jotka kykenevät lievittämään oireita, hidastamaan taudin kulkua ja ehkäisemään (tai ainakin lykkäämään) vakavimpia seurauksia.
MAHDOLLISET Oireelliset hoidot
Luettelossa mahdollisista oireenmukaista hoitoa osteogenesis imperfecta, seuraavat erottuvat:
- Kynsien kirurginen lisäys pisimpien luiden sisälle (Huom. Tätä toimintoa kutsutaan sauvaus intramedullaarinen;
- Murtumien ja / tai luun epämuodostumien konservatiivinen tai kirurginen hoito;
- Hammashoito hampaiden terveyden turvaamiseksi;
- Kipua lievittävät hoidot, jos kyseessä ovat erittäin tuskalliset useat murtumat;
- Fysioterapia, lihasten pidentämiseen ja vahvistamiseen.Elastisen ja vahvistavan lihaskoneen avulla voit estää putoamisia, jotka voivat johtaa erilaisiin luunmurtumiin;
- Liikkumisen apuvälineiden käyttö, mukaan lukien pyörätuolit, olkaimet, kainalosauvat jne.
LIIKKEEN EDUT
Henkilöt, joilla on osteogenesis imperfecta, lääkärit suosittelevat jatkuvaa harjoittelua ja liikuntaa yleensä, koska molemmat nämä toiminnot edistävät luuston ja lihasten vahvistumista.
Suositeltavia urheilulajeja ovat: uinti, koska se on "vähävaikutteinen fyysinen toiminta luustolle", ja kävely.
HYÖDYT TERVEESTÄ ELÄMÄSTÄ
Terveen elämän johtamisella, tupakoinnin välttämisellä, liiallisen alkoholin juomisella, liiallisella ja huonolla syömisellä jne. On enemmän kuin erillisiä terveyshyötyjä potilaille, joilla on osteogenesis imperfecta, koska se hidastaa taudin etenemistä ja vähentää luun haurautta.
Oireelliset hoidot kokeiluvaiheessa
Tällä hetkellä lääkärit ja tutkijat arvioivat joidenkin oireenmukaisten hoitojen tehokkuutta, mukaan lukien kasvuhormonihoito ja bisfosfonaattipohjainen suonensisäinen ja oraalinen hoito.
Tällä hetkellä edellä mainittujen tutkimushoitojen tulokset lupaavat hyvää koko lääketieteelliselle yhteisölle.
Ennuste
Osteogenesis imperfecta on sairaus, jonka ennuste on negatiivinen, koska se on parantumaton, heikentää rajusti elämänlaatua ja joissakin tapauksissa aiheuttaa potilaan ennenaikaisen kuoleman.
On kuitenkin huomattava, että myös nykyaikaisten oireenmukaisten hoitojen ansiosta monet ihmiset, joilla on lievä osteogenesis imperfecta, voivat elää miellyttävää ja tyydyttävää elämää.
Ehkäisy
Valitettavasti tällä hetkellä ei ole ennaltaehkäisevää toimenpidettä osteogenesis imperfectaa vastaan.